差分発現解析(DEG)でめぼしい遺伝子を片っ端に並べても、結局「何がどう動いているのか」が見えない…。そんな悩みに刺さるのが、WGCNA(Weighted Gene Co-expression Network Analysis)です。
WGCNAは、サンプル間で「似た発現挙動を取る遺伝子群(=モジュール)」をまとめ、各モジュールを モジュール固有値(Module Eigengene; ME) という1次元の代表値で要約します。あとはMEと表現型(trait)の相関を見るだけで、どの遺伝子群が、どの条件で、どう動いているか が一目でわかる、という仕掛けです。
この記事では、GEOで公開されている GSE186294(EPO投与試験のIllumina RNA-seqデータ、50サンプル、5時点×10被験者)を題材に、edgeR前処理 → モジュール検出 → Trait相関 → ハブ遺伝子抽出 → 機能アノテーション → GSVA整合 → Cytoscape書き出しまでをRコード付きで一気通貫で走らせます。
- edgeR + voom で WGCNA 用の発現行列を準備できる
pickSoftThresholdで β を選び、blockwiseModulesでモジュール検出ができる- kME / kWithin / GS の役割を理解し、統合スコアでハブ遺伝子をランキングできる
- clusterProfiler で GO / KEGG / Reactome / WikiPathway 解析ができる
- ME(PC1基準)と GSVA(サンプル内基準)を使い分けられる
part 1 / part 2 では「WGCNA で何が見えるか」という結果と解釈 を中心にお見せしました。本記事はそれを 自分のデータで同じように再現できる R コード付きの実装ガイド としてまとめたものです。
関連記事WGCNA解析 part 1:RNA-seqデータを使って、疾患に絡む遺伝子ネットワークを可視化する →
関連記事WGCNA解析 part 2:疾患関連モジュールの機能を読み解き、ターゲット候補遺伝子を絞り込む →
part 1 / part 2 を読んで 「同じ解析を自分のデータでもやってみたい」「コード一式が欲しい」 と思った方は、ぜひ本記事を活用してください。
本記事は前半が無料、後半が有料の構成です。
- 無料パート(〜モジュール検出まで) — 環境構築、データ取得、edgeR + voom 前処理、softPower 推定、
blockwiseModulesでモジュール検出までを R コード付きで全公開 - 有料パート(モジュールの読み解き〜Cytoscape まで) — Module-Trait 相関、kME / kWithin / GS の統合スコアによるハブ遺伝子ランキング、GO / KEGG / Reactome / WikiPathway での機能アノテーション、GSVA との整合チェック、Cytoscape 用エクスポートまで
前半だけでも「自分のデータで WGCNA を走らせてデンドログラムが出る」ところまでは確実に到達できます。後半は 「モジュールから主役遺伝子をデータドリブンで絞り込む」一気通貫のスクリプトと解釈 をまとめたパートです。
こんな方におすすめ
- バルクRNA-seqのDEG解析は経験あるが、WGCNAは初めての方
- WGCNAは触ったことがあるが、kME / kWithin / GS の統合や有意モジュールの絞り込みで迷っている方
- 時系列・縦断デザイン(Base → 介入 → Post)でモジュール挙動を可視化したい方
環境とデータを準備する
作業ディレクトリの構成
本記事のコードは、すべて script/ ディレクトリ内のスクリプトから実行する前提で、相対パス ../data/ ../result/ に読み書きします。あらかじめ以下の構成を作っておくとスムーズです。
WGCNA/
├── data/ # 入力データ(GEO から取得 → 整形済みカウント/メタを置く)
├── script/ # 実行スクリプト(本記事のコードを置く)
└── result/
└── wgcna_out/
├── figure/ # softPower・ヒートマップ等の図
├── enrich/ # GO / KEGG / Reactome / WikiPathway
└── network/ # Cytoscape 用エッジ・ノードR から一括で作るなら次のスニペットで OK です(script/ ディレクトリで実行する想定)。
dirs <- c(
"../data",
"../result/wgcna_out/figure",
"../result/wgcna_out/enrich",
"../result/wgcna_out/network"
)
invisible(lapply(dirs, dir.create, recursive = TRUE, showWarnings = FALSE))環境を準備する
本記事は次の環境で動作確認しています。
R.version.string
# "R version 4.5.1 (2025-06-13)"パッケージのインストール
使うパッケージは CRAN 系 と Bioconductor 系 の両方にまたがります。初回は次のように一括インストールしておきましょう。
# まず pacman と BiocManager を入れる
install.packages(c("pacman", "BiocManager"))
# CRAN パッケージ
install.packages(c(
"tidyverse", "matrixStats", "patchwork",
"gt", "gtExtras", "data.table", "igraph",
"msigdbr", "tidymodels"
))
# Bioconductor パッケージ
BiocManager::install(c(
"GEOquery", "edgeR", "WGCNA", "biomaRt",
"clusterProfiler", "org.Hs.eg.db", "ReactomePA", "enrichplot",
"GSVA"
))Bioconductor パッケージは Bioconductor 本体のバージョンと R のバージョンが揃っている必要があります。エラーが出るときは BiocManager::valid() でズレを確認してください。
パッケージのロード
pacman::p_load(
tidyverse, GEOquery, edgeR, WGCNA, matrixStats,
patchwork, gt, gtExtras, data.table, igraph, biomaRt,
clusterProfiler, org.Hs.eg.db, ReactomePA, enrichplot,
msigdbr, GSVA, tidymodels
)WGCNAは内部で allowWGCNAThreads() を呼ぶと並列計算が効きます。データセットが大きい場合は最初に1回だけ実行しておきましょう。
WGCNA::allowWGCNAThreads()- CRAN 配布の R パッケージ(v1.74, 2026-01)。重み付き共発現ネットワーク解析の事実上の標準
- 強相関を β乗で強調する「スケールフリー」な隣接行列を構築し、TOM ベースで遺伝子をモジュール化
- モジュール固有値(ME)で次元圧縮し、表現型との相関やハブ遺伝子探索に展開できる
GSE186294 からデータを取得する
GSE186294 はエリスロポエチン(EPO)投与前後の血液 RNA-seq データセットです。今回はIlluminaプラットフォーム(GPL18573)のサンプル50件を使います。
ill_url <- "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE186294&file=GSE186294_Illumina_RNAseq_raw_counts_EnsemblID.txt.gz&format=file"
dest_gz <- "../data/GSE186294_Illumina_counts.txt.gz"
download.file(ill_url, destfile = dest_gz, mode = "wb")
counts <- read.table(gzfile(dest_gz), check.names = FALSE, sep = " ")メタデータは GEOquery::getGEO() で取得し、GSMごとの characteristics_ch1 をパースします。
gse <- getGEO("GSE186294", GSEMatrix = FALSE)
gsms_all <- GSMList(gse)
is_illumina <- vapply(gsms_all, function(g) Meta(g)$platform_id == "GPL18573", logical(1))
gsms <- gsms_all[is_illumina]
meta <- bind_rows(lapply(gsms, function(g){
ch <- Meta(g)$characteristics_ch1
tibble(
gsm = Meta(g)$geo_accession,
title = Meta(g)$title,
age = sub("^age \\(year\\):\\s*", "", ch[grepl("^age", ch)]),
sex = sub("^Sex:\\s*", "", ch[grepl("^Sex", ch)]),
time = sub("^time \\(day\\):\\s*", "", ch[grepl("^time", ch)])
)
}))
# "EPO4 (16)" → time_label="EPO4", time_day=16
meta <- meta %>%
tidyr::extract(time, into = c("time_label","time_day"),
regex = "([A-Za-z0-9]+) \\(([-0-9]+)\\)", remove = TRUE) %>%
mutate(time_day = as.integer(time_day),
age = as.integer(age),
sample_no = as.integer(str_extract(title, "\\d+")))サンプル列のラベルをGSMアクセッションに揃えて保存します。
colnames(counts) <- meta$gsm
counts$gene_id <- rownames(counts); rownames(counts) <- NULL
counts <- counts |> dplyr::select(gene_id, everything())
write_tsv(counts, "../data/counts.tsv")
meta_out <- meta %>%
transmute(sample = colnames(counts)[-1],
time_label = factor(time_label, levels = c("Base1","Base2","EPO3","EPO4","Post7")),
time_day, age, sex)
write_tsv(meta_out, "../data/meta.tsv")time_label は Base1 / Base2 / EPO3 / EPO4 / Post7 の5水準で、EPO投与前2点・投与中2点・投与後1点の縦断デザインです。meta.tsv の先頭は次のような形になります。
| sample | time_label | time_day | age | sex |
|---|---|---|---|---|
| GSM5643250 | Base1 | −20 | 26 | Male |
| GSM5643251 | Base2 | −4 | 26 | Male |
| GSM5643252 | EPO3 | 2 | 26 | Male |
| GSM5643253 | EPO4 | 16 | 26 | Male |
| GSM5643254 | Post7 | 42 | 26 | Male |
各被験者(10名)について同じパターンで5時点のサンプルが並ぶ縦断デザインで、計50サンプルになります。
edgeR + voom で正規化し、上位変動遺伝子を抽出する
WGCNAに入れる前に「低発現遺伝子のフィルタ」「ライブラリ間正規化」「分散安定化」が必須です。edgeR(TMM正規化)→ voom(log-CPM+分散安定化)の定石でいきます。
counts <- data.table::fread("../data/counts.tsv") |> as.data.frame()
rownames(counts) <- counts$gene_id; counts$gene_id <- NULL
meta <- data.table::fread("../data/meta.tsv") |> as.data.frame()
rownames(meta) <- meta$sample; meta$sample <- NULL
meta <- meta[match(colnames(counts), rownames(meta)), ]
dge <- DGEList(counts = counts)
keep <- rowSums(cpm(dge) > 1) >= ceiling(0.3 * ncol(counts)) # 30%サンプルでCPM>1
dge <- dge[keep, , keep.lib.sizes = FALSE]
dge <- calcNormFactors(dge, method = "TMM")
v <- voom(dge, plot = FALSE)WGCNAは「全遺伝子を入れる」よりも 分散の大きい上位数千遺伝子に絞る ほうがモジュールが綺麗に出ます。今回は上位5,000遺伝子。
topN <- 5000
rv <- matrixStats::rowVars(v$E)
sel <- order(rv, decreasing = TRUE)[seq_len(min(topN, length(rv)))]
datExpr <- t(v$E[sel, , drop = FALSE]) # WGCNAは samples × genes最後にQC。NA や分散ゼロのサンプル/遺伝子があれば落とします。
gsg <- WGCNA::goodSamplesGenes(datExpr, verbose = 3)
if (!gsg$allOK) {
datExpr <- datExpr[gsg$goodSamples, gsg$goodGenes, drop = FALSE]
meta <- meta[rownames(datExpr), , drop = FALSE]
}Trait 行列を構築する
WGCNAの強みは、後段の モジュール-Trait相関 にあります。そのために traits_num(サンプル × trait の数値行列)をここで作っておきます。time_label は5水準の1-hot、それ以外は連続値です。
meta$time_label <- factor(meta$time_label,
levels = c("Base1","Base2","EPO3","EPO4","Post7"))
X_time <- model.matrix(~ 0 + time_label, meta)
colnames(X_time) <- sub("^time_label", "", colnames(X_time))
sex_num <- as.numeric(factor(meta$sex)) - 1
traits_num <- cbind(
X_time,
time_day = as.numeric(meta$time_day),
age = as.numeric(meta$age),
sex = sex_num
)
rownames(traits_num) <- rownames(meta)データの形と前提が整いました。ここからは WGCNA を実際に走らせて、まずは モジュールが検出されるところまで を一気に進めます。
WGCNA を走らせる:モジュール検出まで
ステップ1:ソフト閾値(softPower)を推定する
WGCNAは遺伝子間のピアソン相関を β乗 することで、強い相関を強調し、弱い相関を消す「スケールフリー」な隣接行列を作ります。このβが softPower です。
pickSoftThreshold() は1〜20の各βで「スケールフリー性の指標 SFT.R.sq」と「平均接続度 mean.k.」を計算してくれます。
SFT.R.sq ≥ 0.8 を満たす 最小の β を採用するのが定石。推定不能(NA)になったら power = 6 を経験的フォールバックに置く。
powers <- 1:20
sft <- pickSoftThreshold(datExpr, powerVector = powers,
networkType = "signed", verbose = 5)
softPower <- sft$powerEstimate
if (is.na(softPower)) softPower <- 6 # 推定できないときのフォールバックsigned ネットワークを選んでいるのは、正の相関と負の相関を区別する ためです。signedなら「同じ向きに動く遺伝子」だけが同じモジュールに入るので、解釈が安定します。
可視化はggplot2で。R²と平均接続度の2枚を並べると、なぜそのβを選んだかが論文に書きやすくなります。
x <- sft$fitIndices
p_r2 <- ggplot(x, aes(Power, SFT.R.sq)) +
annotate("rect", xmin = -Inf, xmax = Inf, ymin = -Inf, ymax = 0.8,
fill = "grey90", alpha = 0.6) +
geom_text(aes(label = Power)) +
geom_hline(yintercept = 0.8, linetype = 2) +
labs(x = "Soft threshold (power)", y = "Scale-free topology R²")
p_mk <- ggplot(x, aes(Power, mean.k.)) +
geom_text(aes(label = Power)) +
labs(x = "Soft threshold (power)", y = "Mean connectivity")
combined <- p_r2 + p_mk
ggsave("../result/wgcna_out/figure/1_softPower.png", combined,
width = 10, height = 4, dpi = 300)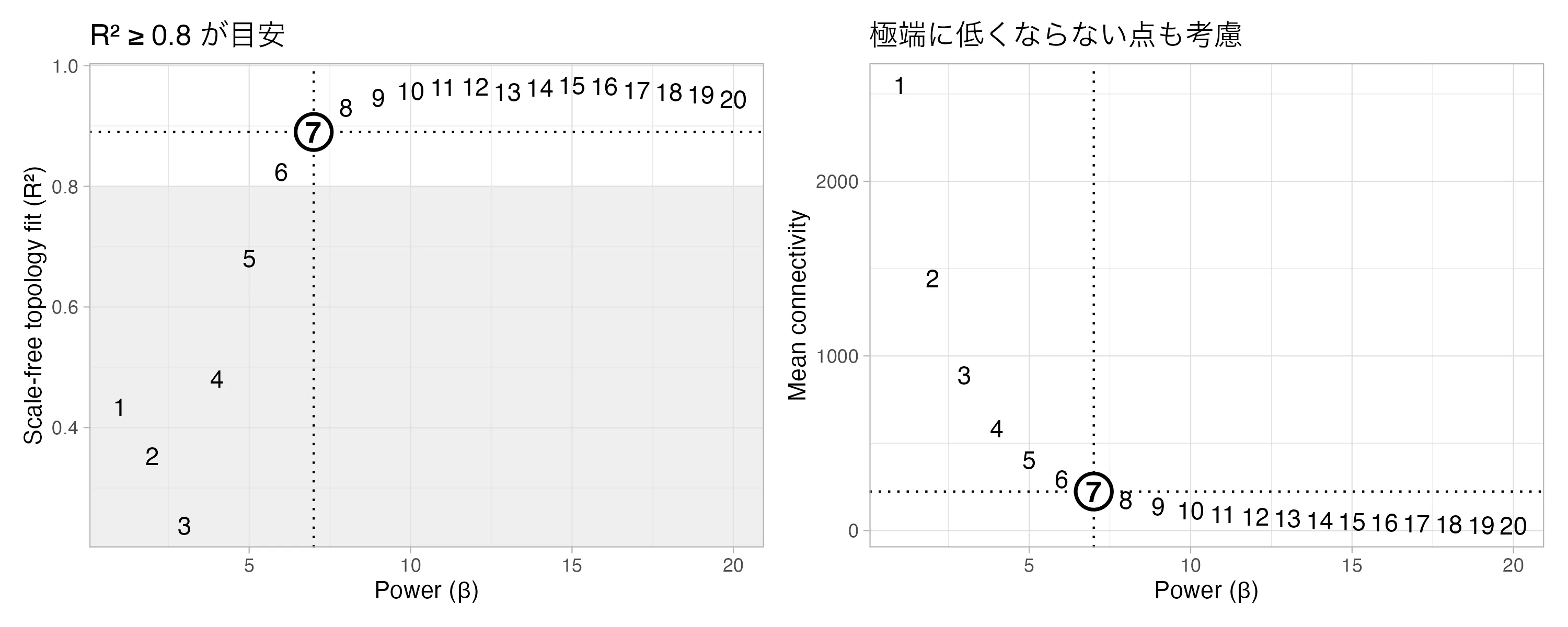 ▲ 左:scale-free R² が 0.8 を超える最小 β は 7。右:β を上げるほど平均接続度は急激に下がる。今回は softPower = 7 を採用。
▲ 左:scale-free R² が 0.8 を超える最小 β は 7。右:β を上げるほど平均接続度は急激に下がる。今回は softPower = 7 を採用。
ステップ2:blockwiseModules でモジュールを検出する
ここがWGCNAの心臓部です。blockwiseModules() を1回呼ぶと、内部で
- 隣接行列の構築
- TOM(Topological Overlap Matrix)の計算
- 階層クラスタリング
- 動的ツリーカットによるモジュール検出
- 類似モジュールのマージ
までを全部やってくれます。
net <- blockwiseModules(
datExpr,
power = softPower,
networkType = "signed",
TOMType = "signed",
minModuleSize = 30, # モジュール最小サイズ
deepSplit = 4, # 細かく分けたいなら大きく(0〜4)
reassignThreshold = 0,
mergeCutHeight = 0.25, # 類似ME間の距離0.25以下をマージ
numericLabels = TRUE,
pamRespectsDendro = FALSE,
saveTOMs = FALSE,
verbose = 3
)
moduleColors <- labels2colors(net$colors)
saveRDS(net, file = "../result/wgcna_out/net_result.obj")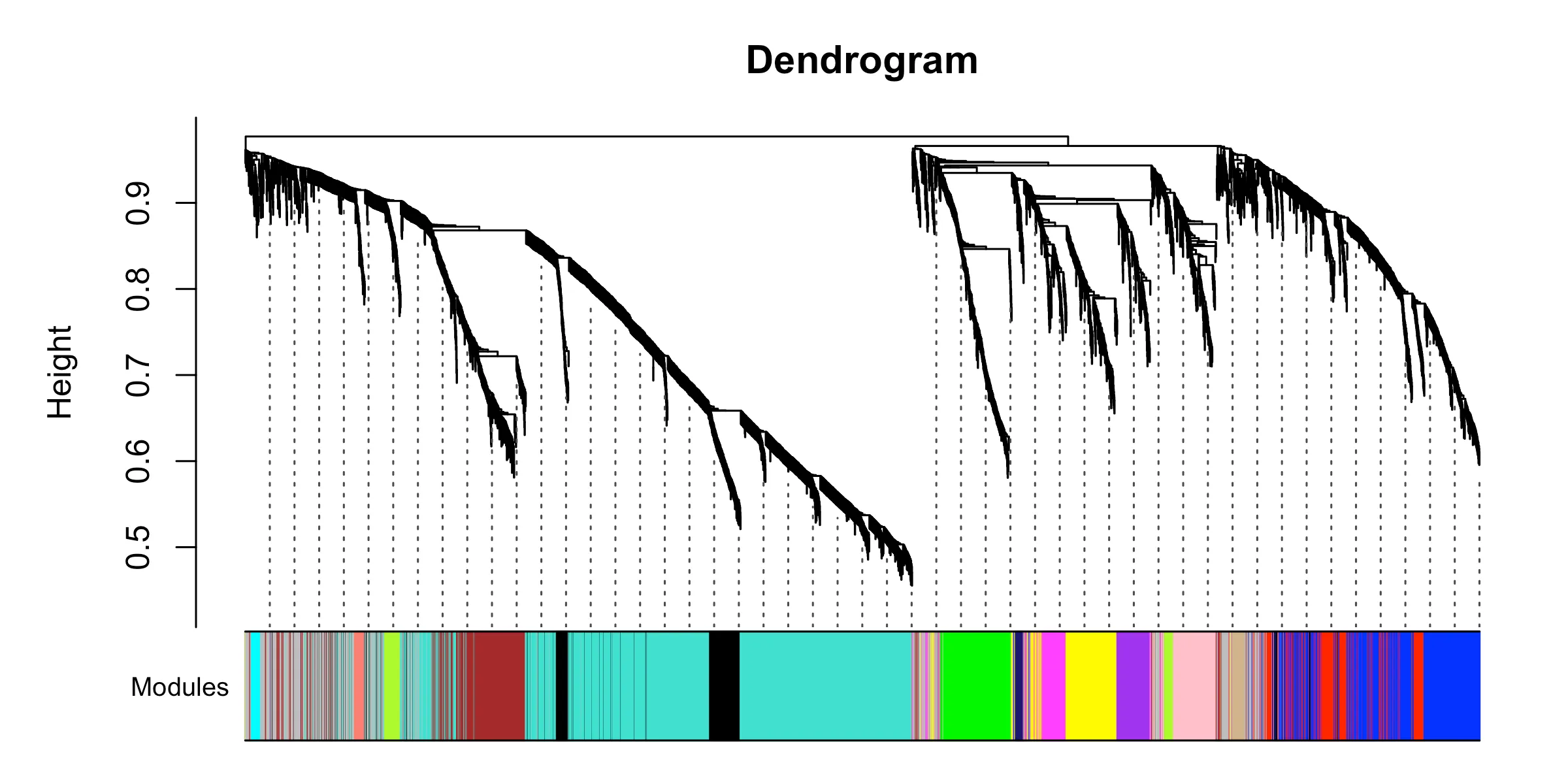 ▲ 階層クラスタリングのデンドログラム下部に、検出されたモジュールが色帯として表示される。色ごとに「似た発現挙動を取る遺伝子の集まり」=モジュールが対応する。
▲ 階層クラスタリングのデンドログラム下部に、検出されたモジュールが色帯として表示される。色ごとに「似た発現挙動を取る遺伝子の集まり」=モジュールが対応する。
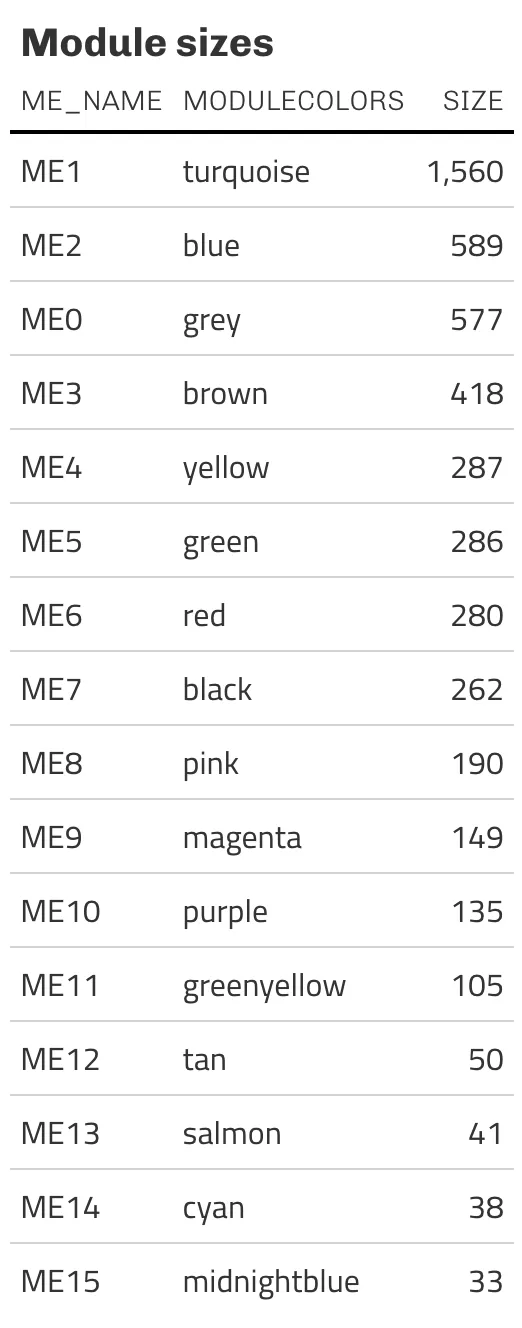 ▲ ME ごとの遺伝子数(モジュールサイズ)。
▲ ME ごとの遺伝子数(モジュールサイズ)。ME0(grey)は「どのモジュールにも属さなかった遺伝子」のゴミ箱なので、解析対象からは除外する。
主要パラメータの目安:
| パラメータ | 設定例 | 効果と調整指針 |
|---|---|---|
minModuleSize | 30 | モジュールの最小遺伝子数。下げすぎると小さなノイズモジュールが乱立する |
mergeCutHeight | 0.25 | ME 同士の距離(1 − 相関)がこの値未満ならマージ。データ量が少ない時は 0.20 まで攻めても良い |
deepSplit | 4 | 0〜4 の整数。大きいほど細かく分割される。迷ったら 2〜4 |
networkType / TOMType | "signed" | 正負の相関を区別。unsigned は絶対値で評価するため解釈が曖昧になりがち |
計算結果は net_result.obj に必ず保存すること。blockwiseModules は遺伝子数が多いと数十分単位で回ることがあり、再計算するハメになるとセッションが消し飛ぶ。
モジュールごとの遺伝子数(サイズ)は次のように一覧化できます。
ME_ref <- WGCNA::orderMEs(WGCNA::moduleEigengenes(datExpr, colors = moduleColors)$eigengenes)
MEs <- WGCNA::orderMEs(net$MEs)
C <- cor(MEs, ME_ref, use = "p")
me_to_color <- setNames(
sub("^ME","", colnames(ME_ref))[max.col(C, ties.method = "first")],
colnames(MEs)
)
moduleColorName <- tibble(
gene_id = colnames(datExpr),
moduleColors = moduleColors
) |>
left_join(tibble(ME_name = names(me_to_color), color = me_to_color),
by = c("moduleColors" = "color"))WGCNA は ME1, ME2, … の数字ラベルと turquoise, blue, brown, … の色ラベルを併用する。実行ごとに対応がズレる ので、上のような変換テーブルを必ず保存しておくこと。後段の図やテーブルの整合がここで決まる。
ここまでで「WGCNAを動かしてモジュールに分ける」までは完了です。ただし、これだけでは 「どのモジュールが、どの表現型と関係していて、その中でどの遺伝子が中心的なのか」 がまだ何もわかりません。
ここから先が本記事の核心 — Trait相関でモジュールに意味を与え、kME / kWithin / GS の統合スコアで ハブ遺伝子を客観的に絞り込み、GO / KEGG / Reactome / WikiPathway で生物学的解釈をつけ、GSVA で整合性をチェックし、最終的に Cytoscape に持っていくまでの「結果を読み解く」工程を、ハマりどころ込みで全部解説します。