普段のRNA-seq等の遺伝子発現解析をだだのDEG解析から一歩進んだ解析としてWGCNA解析を紹介したいと思います。
コードは載せず、WGCNAの流れが分かるような内容にします。
※ コード付きの完全版を別記事で公開しました(前半無料)。実際にRで動かしてみたい方はこちらから。
関連記事【前半無料|全Rコード付き】WGCNA完全パイプライン:WGCNAから主役遺伝子をデータドリブンで絞り込む →🔒 一部有料
WGCNAって?
WGCNA (Weighted Gene Correlation Network Analysis)とは大量の遺伝子発現データから「一緒に上下する(共発現)」遺伝子群=モジュールを見つけ、モジュールと表現型(群・連続値など)の関連を調べ、ハブ遺伝子(モジュールの中心)を仮説として挙げる手法です。因果ではなく相関ベースの探索です。
なお日本語では「WGCNA解析」のほか「WGCNA分析」と表記されることもありますが、同じものを指します。
流れ(ざっくり)
-
前処理:正規化・低発現除去・バッチ補正、外れ値サンプル除去
-
ネットワーク:相関を**softPower(β)**で重み付け(近いほど強い)→TOM(トポロジカル重み)→階層クラスタ→モジュール抽出
-
要約:各モジュールの第1主成分=モジュール固有ベクトル(ME)
-
解釈:MEと形質の相関で“どのモジュールがどの表現型と動くか”を見る
-
優先度付け:kME(モジュール所属度)やkWithin(モジュール内連結度)、GS(Gene Significance:遺伝子と形質の相関)でハブ候補を選ぶ(高kMEかつ高GSが狙い目)
始める前に:WGCNAに必要なサンプル数は?
WGCNAは「サンプル間で遺伝子がどう一緒に動くか(共発現)」を相関で捉える手法なので、相関を安定して推定できるだけのサンプル数が要ります。少なすぎると相関がブレて、出てくるモジュールも信用できません。
公式FAQ(Langfelder & Horvath)の目安は明確です。
-
15サンプル未満では非推奨(“We do not recommend attempting WGCNA on a data set consisting of fewer than 15 samples”)
-
実用的には 20サンプル以上あると安定しやすい
-
基本的に多いほど良い
つまり3 vs 3 のような小規模デザインはWGCNA向きではありません。その場合は素直にDEG解析を使うか、少数サンプル向けの手法を検討した方が無難です。今回扱うGSE186294は、時系列で複数の時点×反復があるため十分な数を確保しています。
「ちょうど何サンプルあれば正解」という厳密な閾値があるわけではなく、15は”これ未満はやめておけ”のライン、20以上で実用、というのが現場感覚です。
WGCNAパッケージの入手(CRAN / Bioconductor)
WGCNAパッケージは CRANで公式配布されています(install.packages("WGCNA"))。ただし依存パッケージの一部(GO.db・impute・preprocessCore など)がBioconductor側にあるため、先に BiocManager でそれらを入れておくとインストールでつまずきません。
インストールから実際の解析コード一式(ハブ遺伝子の絞り込みまで)は、全Rコード付きの完全版にまとめてあります。
関連記事【前半無料|全Rコード付き】WGCNA完全パイプライン →🔒 一部有料
今回扱うデータに関して
今回使うデータセットは「GSE186294」にしました。
被験者は4週間にわたり、2日おきに体重1kgあたり50IUの組換えヒトエリスロポエチン(rHuEPO)注射を受けた。rHuEPO投与期間中(4週間)、毎日経口鉄剤(元素鉄100mg、硫酸第一鉄錠、Almus社製、英国バーンスタブル)が投与された。ここで、ベースライン時(Base1およびBase2)、rHuEPO投与期間中(初回投与後2日目および2週間後;EPO3およびEPO4)、ならびにrHuEPO投与終了後2週間(Post7)に採取した全血サンプルを、イルミナNextSeq 500を用いて解析した。
Base 1 → Base 2 → EPO3 → EPO4 → Post 7 という時系列のサンプルです。
WGCNA解析実行結果
1. softPower(β)の設定
softPower(β)とは?
まずは**softPower(β)**を設定します。
各遺伝子の相関cor(i, j)を計算した後、この相関に掛ける重****みのことです。
βを上げるほど、強い相関だけが残りやすく(スパース化)、弱い相関は0に近づきます。
unsigned:
signed: (負相関は小さくなる)
この重みを使って TOM → 階層クラスタ → モジュール が決まります。
なお、ここで決まるモジュールの数やサイズは blockwiseModules() の引数(deepSplit・minModuleSize・mergeCutHeight など)で大きく変わります。パラメータで詰まったときは、こちらの番外編が逆引きに使えます。
関連記事WGCNA blockwiseModules の主要パラメータ:deepSplit・minModuleSize・mergeCutHeight の意味と決め方 →
βはどうやって決める?
それではβを決めていきます。
WGCNAでは、遺伝子ノード次数分布がスケールフリー(べき乗則)に近くなるようにβを定める。
WGCNAのpickSoftThreshold()関数を実行し複数のβを試し、以下の2つを検討する:
-
Scale-free fit(R²):べき乗則にどれだけフィットするか
-
Mean connectivity:平均連結度(βを上げると下がる)
基本ルール:
-
最小のβでR² ≳ 0.8(小規模やノイジーなら 0.7 も妥協点)
-
かつ mean connectivity が極端に低くなり過ぎない地点を選ぶ
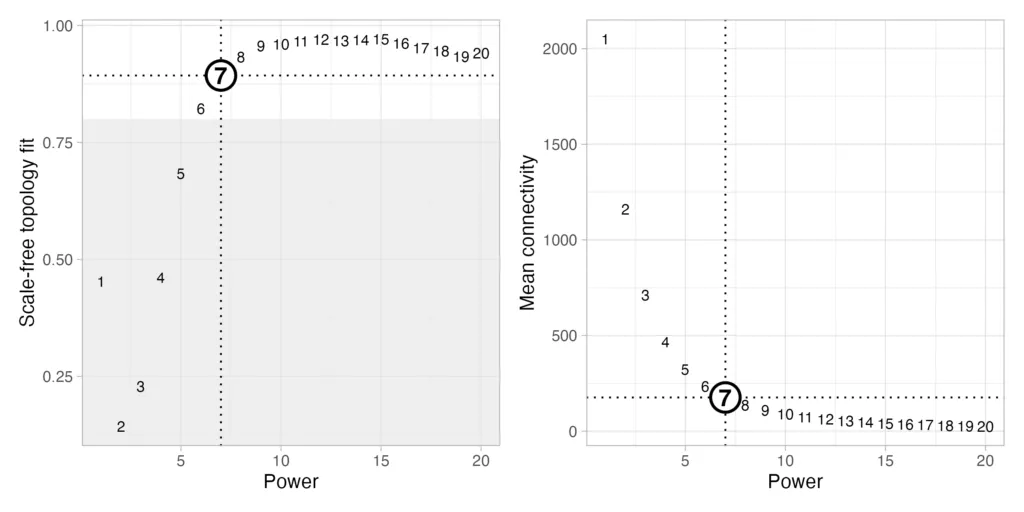
今回はβ=7 が選択されました。
系統樹の確認
は既出ですので省きます。
はノード(遺伝子)iの重み付き次数
TOMの分子の左側 は、
遺伝子iと遺伝子jがその他の遺伝子とどれぐらい共通で繋がっているかの指標です。
次は分母の説明ですが、
これは理論上取りうる最大の重なりで割って 0 - 1に正規化しています。
は距離に変換しています。TOMが近さだから、逆にすることで遠さ=距離に変換しているわけです。
これをクラスタリングや樹形図を作る際には距離を使用するためこの変換が必要です。
その結果が以下の樹形図です。
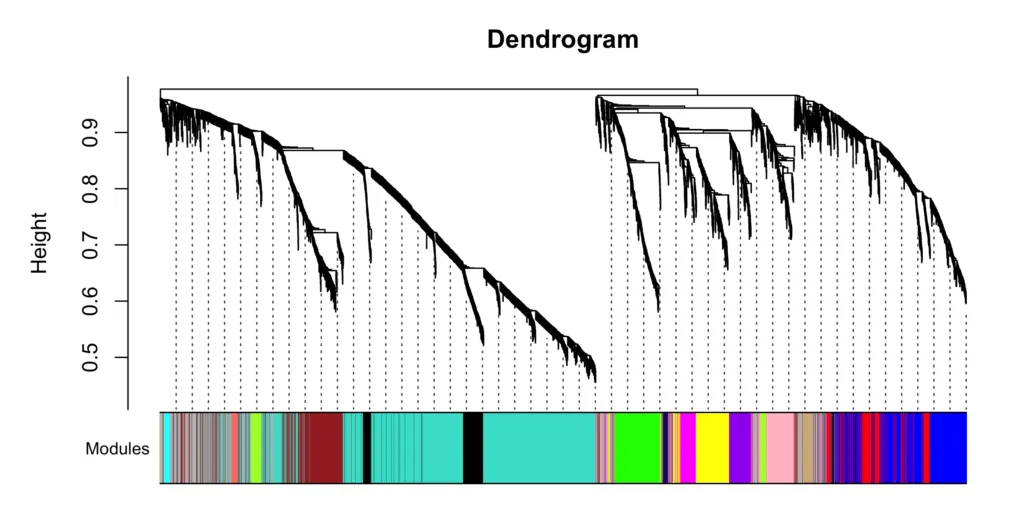
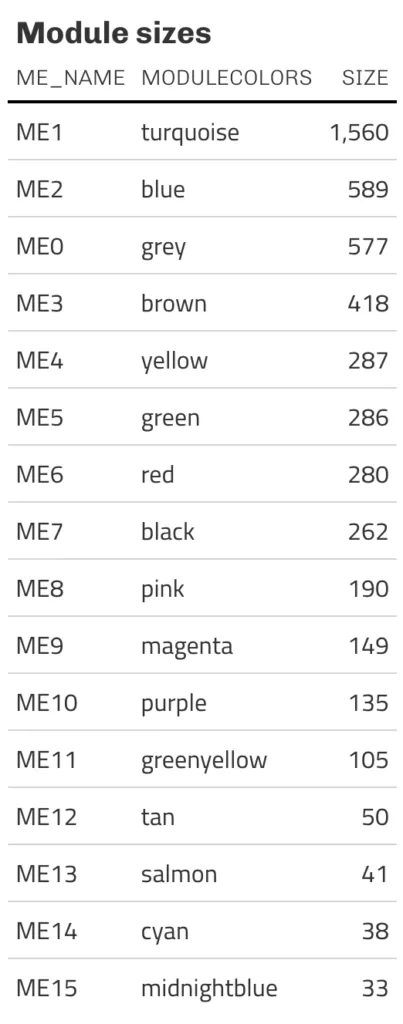
また各モジュールのサイズ(遺伝子数)は以下の通り
また、各モジュールのmodule eigengenes (ME)を算出し、モジュール間の類似性を比較しています。
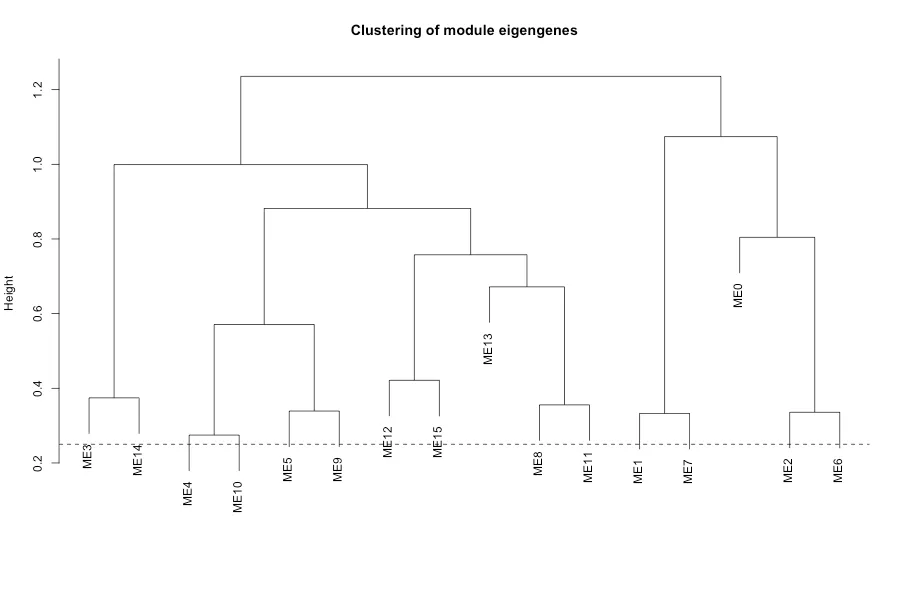
MEとは各モジュールの遺伝子発現を主成分分析したときの第1主成分=サンプル方向の代表発現(モジュール活動度)
1モジュール1サンプルにつき1つの値になる
着目モジュールの選定
次は、作成したモジュールのうち、どのモジュールに着目するかを決めていきます
どのように着目するモジュールを選んでいくかには、都度変わってくるかと思いますが、今回は各モジュールと各処置との関係から選んでいこうと思います。
各サンプルの各モジュールの「値」が処置(Base, EPO, Postなど)に伴って変動するどうかを検討すれば良いので、サンプルラベルと各サンプルのモジュールの「値」の相関を見ていきます。
この各サンプルのモジュールの「値」にMEを使います。
その結果が以下です。
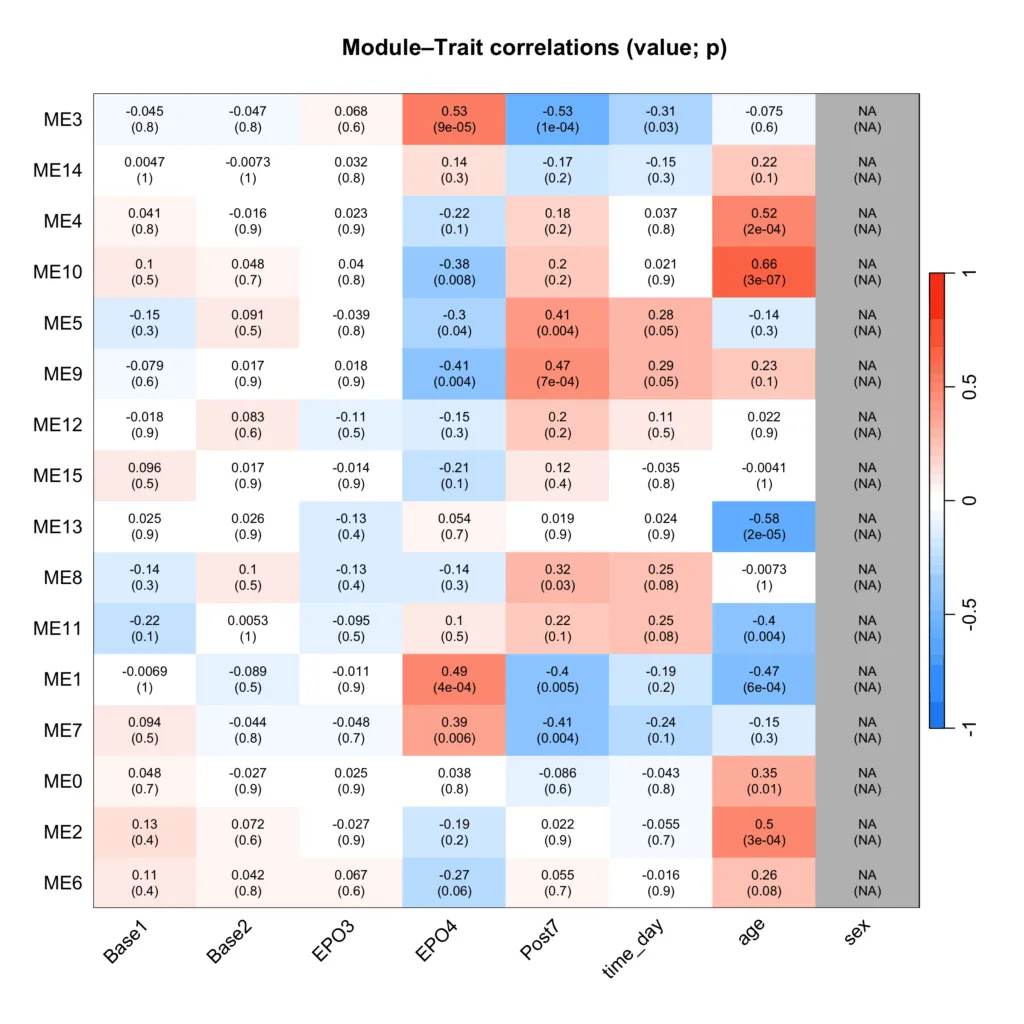
まずは、EPO4と高い相関が見られるModule 3に着目していきたいと思います。
今回はココまで、次回からModule 3について詳しく解析していきたいと思います。
関連記事WGCNA解析 part 2:疾患関連モジュールの機能を読み解き、ターゲット候補遺伝子を絞り込む →
同じ解析を自分のデータでやってみたい方へ — このシリーズの実装記事に、GEOデータ取得 → edgeR/voom 正規化 → モジュール検出 → kME / kWithin でのハブ遺伝子の絞り込み → Cytoscape まで、全Rコードとハマりどころをまとめています。前半無料なので、まず動かして試せます(後半 ¥500)。